RNA-Seq: Dari reads ke ekspresi gen
Oleh Agus Wibowo

Daftar isi
- Apa itu studi RNA-seq?
- Tentang data yang digunakan dan persiapannya
- Quality control data sequencing
- Memilih tools bioinformatika
- Instalasi tools untuk RNA-Seq
- Menyiapkan index referensi dengan HISAT2
- Alignment dan konversi dari SAM ke BAM
- Merakit transkrip dan menghitung level ekspresi gen menggunakan StringTie
- Analisis perbedaan ekspresi
- Analisis fungsi gen - GO dan KEGG pathway
- Apa yang perlu dipertimbangkan sebelum studi RNA-seq
Catatan: Jika Anda mengikuti tutorial ini dari awal hingga akhir, perlu diketahui bahwa proses alignment dan kuantifikasi ekspresi memerlukan sumber daya komputasi yang cukup dan waktu yang tidak singkat. Sebagai gambaran, ketika saya menjalankan proses ini di laptop dengan RAM 12 GB dan prosesor Intel i5 gen 10 pada LINUX UBUNTU, ini memerlukan waktu sekitar 4 jam. Sehingga, tutorial ini mungkin masih achievable.
Pastikan juga Anda memiliki ruang penyimpanan data yang cukup, minimal 50 - 100 GB disertai dengan koneksi internet yang stabil untuk mendownload data FASTQ.
Namun jangan khawatir, bagi Anda yang tidak memiliki akses ke komputer dengan spesifikasi tinggi, saya telah menyediakan file hasil preproses dalam format
*.rdsyang siap dianalisis, tapi setidaknya Anda memahami workflow umum dalam studi RNA-seq.Dalam tutorial ini, kita akan menggunakan bahasa pemrograman Bash dan R dalam lingkungan Linux. Anda bisa menggunakan VS Code baik untuk mengedit skrip maupun menjalankannya melalui terminal secara langsung. Script bash dan R untuk menangani data dalam tutorial ini sudah saya sediakan.
Apa itu studi RNA-Seq?
Menyelami percakapan genetik
RNA sequencing (RNA-seq) adalah teknologi sekuensing generasi baru yang memungkinkan kita “mendengar” aktivitas gen dalam sel untuk mengetahui gen mana yang aktif, seberapa besar ekspresinya, dan bagaimana ekspresi itu berubah dalam berbagai kondisi biologis. Tidak seperti microarray yang hanya mendeteksi gen yang telah diketahui sebelumnya, RNA-seq memetakan seluruh transkriptom (semua RNA yang aktif dalam sel pada waktu tertentu), termasuk RNA langka dan varian splicing baru yang belum pernah teridentifikasi.
Teknologi ini telah merevolusi biologi molekuler dengan menghadirkan gambaran menyeluruh tentang dinamika ekspresi gen, baik pada level jaringan maupun, lebih baru lagi, pada level sel tunggal melalui pendekatan single-cell RNA-seq. Pendekatan ini membuka jendela terhadap keragaman seluler yang sebelumnya tersembunyi, memungkinkan identifikasi tipe sel baru, jalur diferensiasi, dan respons sel spesifik terhadap lingkungan.
Mengapa penting untuk akuakultur?
Dalam konteks akuakultur, RNA-seq menjadi alat yang strategis dan canggih untuk memahami bagaimana respon organisme budidaya terhadap suatu kondisi tertentu, misalnya stres, penyakit, pakan, serta kondisi lingkungan secara molekuler. Sehingga kita dapat mengungkap gen-gen yang terlibat dalam imunitas, pertumbuhan, metabolisme, hingga adaptasi terhadap budidaya intensif.
Penerapannya mencakup identifikasi biomarker untuk deteksi dini penyakit, seleksi genomik berbasis ekspresi gen, optimalisasi pakan, pengembangan strain unggul, hingga penerapan gene editing (CRISPR). Misalnya, dengan menggunakan single-cell RNA-seq, kita bisa mengeksplorasi peran masing-masing sel dalam organ penting seperti hati, usus, insang atau organ lain untuk menghadirkan wawasan yang jauh lebih tajam untuk manajemen kesehatan dan nutrisi.
Dari gen ke keputusan produksi
RNA-seq menjembatani dunia molekuler dan praktik budidaya. Dengan memahami “bahasa” genetik secara mendalam, kita tak hanya menjawab pertanyaan biologis, tetapi juga membuat keputusan produksi yang lebih presisi, efisien, dan berkelanjutan. RNA-seq bukan sekadar teknologi, melainkan fondasi untuk membangun akuakultur yang cerdas, yang tidak lagi menebak, tapi memahami langsung apa yang sebenarnya dibutuhkan oleh organisme budidaya.

Sumber gambar: Liu et al 2024
Tentang data yang digunakan dan persiapannya
Studi analisis ekspresi gen pada ikan Turbot
Dalam tutorial ini, kita menggunakan data dari studi oleh Lange et al. (2014) yang meneliti tentang bagaimana perbedaan ekspresi gen pada udang vanamei (Liptopenaeus vannamei) pasca terinfeksi oleh toxin Pir A/B dari bakteri Vibrio sp yang umumnya menyebabkan penyakit acute hepatopancreatic necrosis disease (AHPND). Penelitian ini sangat relevan dan penting karena sering kali AHPND menyebabkan kematian massal pada udang budidaya sebelum 30 hari pemeliharaan.
Penelitian ini menganalisis perubahan ekspresi gen pada sampel hepatopankreas menggunakan RNA-seq berbasis short reads sequencing (Illumina HiSeq X Ten - paired-end reads). Sehingga dengan memahami bagaimana respon molekuler terutama ekspresi gen bekerja ketika udang terinfeksi, ini akan membantu dalam pengembangan strategi pencegahan yang lebih efektif, seperti seleksi genetik untuk ketahanan penyakit, pengembangan vaksin, atau pengelolaan lingkungan yang lebih tepat dalam sistem budidaya.

Sumber gambar: Nusantics
Persiapan data hasil sekuensing
Dataset lengkap dapat diakses melalui ENA (European Nucleotide Archive) dengan kode bio project PRJNA823051. Untuk melihat detail eksperimen, kita bisa klik pada bagian “Sample Accession”, di sana terdapat informasi seperti perlakuan dan organ yang digunakan.
Pada database, kita akan mengunduh semua data performat *.fastq.gz (sekitar 86 GB). Untuk memudahkan, Anda dapat mengklik tombol “Download All” pada bagian “Generated FASTQ files: FTP”. Tindakan ini akan mengunduh sebuah file .sh, yaitu skrip otomatis yang dapat dijalankan menggunakan wget untuk mengunduh semua file paired-end reads FASTQ secara langsung.
Setelah mengunduh file .sh, jalankan perintah bash berikut satu persatu di terminal.
#bash
# buat folder tempat menyimpan data fastq.gz
mkdir -p raw_reads
# pindahkan script .sh ke raw_reads
mv file_script_download_ena.sh raw_reads/file_script_download_ena.sh
# download semua file fastq.gz
bash raw_reads/file_script_download_ena.sh
Tunggu proses pengunduhan data, mungkin akan memakan waktu cukup lama tergantung kecepatan internet yang Anda miliki, “so, be chill and take your coffee. Setelah selesai, semua file *.fastq.gz akan berada di dalam folder raw_reads.
Perhatikan penamaan filenya:
SRR: singkatan dari Sequence Read Run yang merupakan ID akses unik (run accession) dari satu unit percobaan sekuensing.- 7 digit angka setelah
SRR: merepresentasikan kode percobaan yang mana 2 angka terakhir merepresentasikan detail perlakuan. _1.fastq.gzdan_2.fastq.gz: menunjukkan bahwa data ini berasal dari paired-end sequencing, artinya satu fragmen DNA dibaca dari dua arah, forward (_1) dan reverse (_2). Semua file pada dasarnya berformat FASTQ, namun dikompresi dengan gunzip.
Memahami penamaan file ini sangat penting karena membantu kita mengelola dan mengidentifikasi data sekuensing dengan benar, terutama saat bekerja dengan banyak sampel.
Menyiapkan reference genome
Selain data reads hasil sequencing, kita juga membutuhkan genom referensi yang valid untuk alignment. Untuk udang vannamei, kita dapat menunduh 2 jenis file dari NCBI dengan kode assembly ASM4276789v1, untuk menunduh dua jenis file referensi yaitu:
- Genom referensi dengan format:
_genomic.fna.gz - Anotasi genom dengan format:
_genomic.gff.gzdan_genomic.gtf.gz
Download kedua data ini dengan menggunakan wget:
# bash
# buat folder untuk menyimpan genom referensi
mkdir -p references
# download genom referensi dan anotasinya dan simpan ke folder references
wget -O references/GCF_042767895.1_ASM4276789v1_genomic.fna.gz https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/042/767/895/GCF_042767895.1_ASM4276789v1/GCF_042767895.1_ASM4276789v1_genomic.fna.gz
wget -O references/GCF_042767895.1_ASM4276789v1_genomic.gtf.gz https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/042/767/895/GCF_042767895.1_ASM4276789v1/GCF_042767895.1_ASM4276789v1_genomic.gtf.gz
wget -O references/GCF_042767895.1_ASM4276789v1_genomic.gff.gz https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/042/767/895/GCF_042767895.1_ASM4276789v1/GCF_042767895.1_ASM4276789v1_genomic.gff.gz
# ekstrak file genom referensi dan anotasi
gunzip references/GCF_042767895.1_ASM4276789v1_genomic.fna.gz
gunzip references/GCF_042767895.1_ASM4276789v1_genomic.gtf.gz
gunzip references/GCF_042767895.1_ASM4276789v1_genomic.gff.gz
Sekarang kita sudah memiliki semua data yang dibutuhkan.
- File reads hasil sequencing yang disimpan di:
raw_reads/*.fastq.gz - Genom referensi dan anotasinya yang disimpan di:
references/
Quality control data sequencing
Inspeksi awal file sequencing
Cara paling sederhana untuk melihat seperti apa hasil sequencing adalah melalui terminal bash.
Pertama, kita bisa lihat terlebih dahulu bagaimana struktur file .fastq.gz menggunakan head.
# bash
# lihat struktur file SRR18604305_2.fastq.gz
gunzip -c SRR18604305_2.fastq.gz | head
Perintah di atas akan menghasilkan 3 sekuen yang kodenya selalu diawali dengan @, di bawahnya secara berturut-turut adalah urutan basa DNA dan skor kualitas:
@SRR18604305.1 1/2
CAGCCTCTGAGCTTCAACGACTACGTTCGCGCCATCGATATTCCCGCTCAGGGTCACGCTGCCTCTGGCGACTGCATCGTTTCCGGCTGGGGCGCTCTCACTGAAGGAGGCTGCTCCCCCAGTGCCCTCCAGAAGGTGTCCGTTCCCATC
+
??????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????
@SRR18604305.2 2/2
ACCGGCGGGGGCCTCCCCCCTGAGCTGCAGCCCCTGGCGGCGCGGGTGCCGGAGGTCAGCAACTCCATCTGCGTGGTCATTGAGTACGAGGACGTGTGGGGGGCGGCGCGCGCCCTGCAGGAGCTGGACGACCCTAAGATGAGCCTGCAC
+
??????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????
@SRR18604305.3 3/2
CCTTAATGACGGCAAACTCTTGGAGCAGAAACACTGGTTCTCCCTTTTCAATACAAGGCATCGTAATGAGGCACTCCTGCTTTTCGACGTCCTCATCCACTGCAAAGACTGTTCATCCTTTGTCGGCAATGCAGCCTACTTCCGTCAGAA
Untuk penjelasan detail tentang format file FASTQ, bisa lihat pada artikel: Mengenal berbagai format file NGS
Kemudian, jika kita ingin mengetahui seberapa panjang setiap reads dan berapa banyak reads yang dihasilkan, kita bisa menggunakan gunzip yang dikombinasikan dengan tail, wc, dan awk.
# bash
# melihat seberapa panjang sekuen untuk 50 reads dan melihat konsitensinya
gunzip -c SRR18604305_2.fastq.gz | head -n 100 | tail -n 1 | wc -c
# melihat ada berapa banyak reads
gunzip -c SRR18604305_2.fastq.gz | awk 'END {print NR/4}'
Secara berturut-turut, output dari perintah di atas akan menghasilkan:
# panjang urutan basa nukleotida
151
# jumlah reads
40425934
Penjelasan perintah:
gunzip -c: membuka file.gztanpa perlu meng-ekstrak nya karena menggunakan option-c.head -n 100: mengambil informasi untuk 50 nukleotida (baris pertama adalah header reads dan baris kedua adalah urutan sekuen nukleotida).tail -n 1: mengambil baris ke-2 saja (urutan basa nukleotida)wc -c: menghitung berapa banyak karakter (banyaknya basa nukleotida)awk 'END {print NR/4}': menghitung total baris (NR - Number of Reads), lalu membaginya dengan 4 (karena setiap read FASTQ terdiri dari 4 baris).- Antar perintah selalu dihubungkan dengan pipe (
|), artinya –> gunakan hasil dari perintah “x” untuk perintah “y”.
Jadi secara umum, proses sequencing paired-ends berhasil membaca sekitar 17 juta reads dengan masing-masing reads berisi 101 basa nukleotida.
Quality control data FASTQ
Sampai tahap ini, kita hanya mengetahui bagaimana struktur hasil sekuensing dan berapa banyak reads yang dihasilkan, tapi belum mengetahui bagaimana kualitas keseluruhan data hasil sequencing dan perlukah data yang diperoleh “diolah” sehingga siap untuk proses berikutnya. Di sinilah tahap quality control (QC) diperlukan.
Sebelumnya, perlu diketahui bahwa setiap data yang di-publish di database seperti ENA atau NCBI-SRA, semuanya adalah data yang “bersih” dan siap analisis, sehingga pada dasarnya kita bisa lewati tahap QC. Namun jika Anda melakukan penelitian RNA-seq dan menerima hasil sekuensing dari Illumina misalnya, proses QC adalah tahapan WAJIB. Untuk mengetahui bagaimana proses ini dilakukan, Anda bisa pelajari pada artikel berjudul: Quality control hasil sequencing.
Memilih tools bioinformatika
Pemilihan tools bioinformatika dalam studi RNA-seq sebaiknya disesuaikan dengan kebutuhan spesifik dari studi dan pertanyaan biologis yang ingin dijawab. Hal ini penting karena hingga saat ini, telah dikembangkan ratusan tools dengan fungsi, kelebihan, dan kekurangan masing-masing, mulai dari tahap preprocessing, alignment, kuantifikasi, hingga analisis diferensial.
Misalnya, jika fokus penelitian hanya screening awal ekspresi gen dengan memprioritaskan kecepatan dan efisiensi komputasi, maka tools pseudo-alignment seperti Salmon atau Kallisto bisa menjadi pilihan yang tepat. Namun, jika studi memerlukan informasi posisi genomik secara akurat, seperti mendeteksi splice junction dan mencari variasi gen baru, maka pendekatan full alignment ke genom seperti STAR, HISAT2, atau Bowtie2 lebih disarankan.
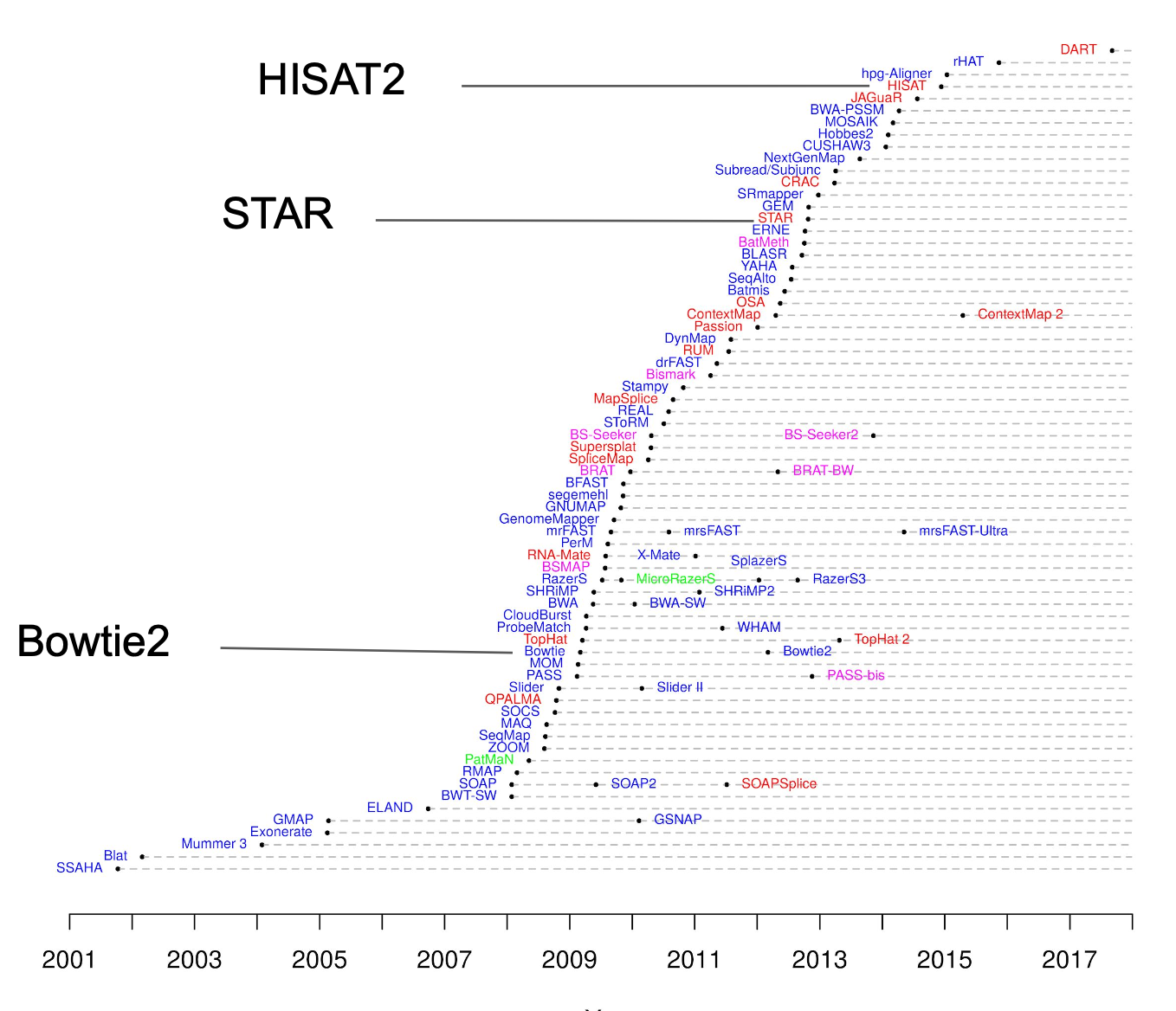
Sumber gambar: bioinformatics.ccr.cancer.gov
Pada tutorial ini misalnya, karena tujuannya adalah ingin mengetahui bagaimana perbedaan ekspresi gen dari dua kondisi (normal vs infeksi), namun pipeline dapat tetap berjalan pada komputer umum (non-HPC), maka HISAT2 sebagai aligner, dan HTSeq sebagai quantifier, dan DESeq2 sebagai differential expression analyzer merupakan kombinasi yang tepat.
Pada tutorial ini, saya juga menyediakan data hasil dari pipeline STAR + RSEM untuk perbandingan. Meskipun pipeline STAR + RSEM membutuhkan sumberdaya komputasi yang lebih tinggi, pipeline ini memberikan estimasi abundance yang akurat untuk transcript yang sudah teranotasi. Saya akan memberikan tutorial khusus untuk pipeline ini pada artikel yang lain.
Perbandingan kedua pipeline ini penting untuk memahami trade-off antara kecepatan komputasi dan kemampuan discovery. Dengan menyajikan kedua pendekatan, tutorial ini memberikan pemahaman praktis tentang bagaimana pilihan tools dapat mempengaruhi hasil analisis dan membantu peneliti menentukan strategi yang sesuai dengan tujuan penelitian mereka. Selain itu, untuk menambah wawasan, anda bisa baca referensi berikut untuk melihat bagaimana perbedaan mungkin dihasilkan ketika kita menggunakan pipeline yang berbeda.
Catatan: Karena proses analisis RNA-Seq cukup panjang dan kompleks, terutama jika dikerjakan untuk banyak sampel satu per satu, di akhir tutorial ini saya sudah menyiapkan script dan konfigurasi yang bisa langsung digunakan. Semua sudah dirancang agar dapat menangani banyak sampel sekaligus menggunakan pipeline: HISAT2 - StringTie - DESeq2, selama data berasal dari paired-end reads, apapun jenis sampelnya.
Instalasi tools untuk RNA-Seq
Dalam tutorial ini, kita membutuhkan 4 tools utama, yaitu; HISAT2 untuk alignment, Samtools untuk konversi file .sam ke .bam, HTSeq untuk kuantifikasi ekspresi, DESeq2 untuk analisis perbedaan ekspresi gen, serta beberapa paket R lainnya.
Karena HISAT2, Samtools, dan StringTie tersedia melalui conda, maka kita dapat menjalankan perintah sebagai berikut.
# bash
# buat dan aktifkan environment baru (opsional namun disarankan)
conda create -n rna_seq -y
conda activate rna_seq
# instalasi tools utama
conda install -c bioconda hisat2 samtools htseq -y
Install paket-paket dari R dengan perintah berikut
# masuk ke R console melalui terminal dengan mengetikkan R
R
Jalankan proses instalasinya satu-persatu mulai dari instalasi paket dari Bioconductor dan dilanjutkan dengan instalasi paket dari CRAN:
# r
# install paket dari Bioconductor
BiocManager::install(c("DESeq2", "limma", "ComplexHeatmap"))
# install paket dari CRAN
install.packages(c(
"tidyverse", "ggrepel", "RColorBrewer", "viridis", "pheatmap",
"VennDiagram", "patchwork", "corrplot", "ggcorrplot",
"grid", "reshape2", "circlize", "stringr", "dplyr"
))
# terakhir, keluar dari console R dengan perintah
q() # y --> Enter
Menyiapkan index referensi dengan HISAT2
Index referensi adalah langkah awal yang penting dalam analisis RNA-seq, di mana komputer membangun “peta navigasi” genom untuk memandu proses alignment (pencocokan fragmen RNA ke lokasi asalnya di DNA). Tanpa index, alignment akan seperti penyusunan puzzle tanpa melihat gambar aslinya. Proses ini mencakup ekstraksi informasi dari anotasi genom (seperti exon dan splice sites), lalu menyusunnya bersama urutan genom menjadi struktur data yang dioptimalkan agar alignment bisa dilakukan secara cepat dan presisi.
Sebelum membuat index, kita perlu mengekstraksi splice site dan daerah exon dari file .gtf. Gunakan perintah berikut.
# bash
# buat direktori kerja HISAT-StringTie
mkdir -p HISAT-StringTie
# ekstrak informasi splice site dan exon dari file .gtf
hisat2_extract_splice_sites.py references/GCF_042767895.1_ASM4276789v1_genomic.gtf > HISAT-StringTie/splice_sites.txt
hisat2_extract_exons.py references/GCF_042767895.1_ASM4276789v1_genomic.gtf > HISAT-StringTie/exons.txt
Kemudian kita bisa membuat index dengan menggunakan hisat2-build:
# bash
# buat direktori untuk menyimpan index di dalam folder HISAT-StringTie
mkdir -p HISAT-StringTie/index
# jalankan indexing
hisat2-build -p 4 \
--ss HISAT-StringTie/splice_sites.txt \
--exon HISAT-StringTie/exons.txt \
references/GCF_042767895.1_ASM4276789v1_genomic.fna \
HISAT-StringTie/index/ASM2237912v1_hisat2_index
Ketika proses pembuatan index selesai, maka di dalam direktori HISAT-StringTie/index/ akan ada 8 file index dengan format .ht2 untuk digunakan sebagai input pada proses alignment.
Alignment dan konversi dari SAM ke BAM
Proses alignment dilakukan untuk semua sampel, dengan input data diantaranya:
- File-file index:
HISAT-StringTie/index/ASM2237912v1_hisat2_indexdengan format.ht2 - Raw data sequencing (paired-end reads): misal kita mengerjakan untuk data
SRR18604305.
Jalankan proses alignment untuk semua sampel dengan perintah dasar sebagai berikut:
# bash
# buat direktori hasil alignment
mkdir -p HISAT-StringTie/aligned_data
# jalankan alignment
hisat2 -p 4 --dta \
-x HISAT-StringTie/index/ASM2237912v1_hisat2_index \
--rna-strandness --rf \
-1 raw_reads/SRR18604305_1.fastq.gz \
-2 raw_reads/SRR18604305_2.fastq.gz \
-S HISAT-StringTie/aligned_data/SRR18604305.sam
Catatan: option
-p 4berarti kita menggunakan 4 core processor, sehingga anda dapat menyesuaikan dengan kapabilitas komputasi yang anda miliki, misal-p 6jika ingin menggunakan 6 core processor untuk alignment yang lebih cepat. Option--rna-strandness --rfdigunakan karena data yang kita kerjakan berasal dari strand-specific (stranded) RNA-seq dengan orientasi reverse-forward (library dibangun dari untai RNA negatif).
Hasil dari perintah di atas adalah file-file dengan format .sam yang berisi hasil alignment, yakni informasi detail tentang bagaimana setiap pasangan fragmen RNA (read pair) dipetakan ke lokasi tertentu di genom referensi. File ini mencatat posisi, orientasi, kualitas pencocokan, dan status splice site dari tiap read, sehingga menjadi dasar penting untuk analisis ekspresi gen selanjutnya. Karena file .sam biasanya berukuran sangat besar (>15 GB), maka ini perlu dikonversi ke format .bam yang lebih ringkas dan efisien untuk diproses lebih lanjut, terutama untuk perakitan transkrip (transcript assembly) menggunakan StringTie.
Konversi file dari .sam ke .bam dapat dilakukan melalui perintah dasar sebagai berikut:
# bash
# buat direktori untuk menyimpan file .bam
mkdir -p HISAT-StringTie/bam_data
# lakukan konversi menggunakan samtools
samtools view -@ 6 -bS HISAT-StringTie/aligned_data/SRR18604305.sam > HISAT-StringTie/bam_data/SRR18604305.bam
# Sorting BAM file
samtools sort -@ 6 HISAT-StringTie/bam_data/SRR18604305.bam -o HISAT-StringTie/bam_data/SRR18604305.sorted.bam
# Indeks BAM file
samtools index HISAT-StringTie/bam_data/SRR18604305.sorted.bam
# Hapus file BAM untuk menghemat ruang penyimpanan
rm HISAT-StringTie/bam_data/SRR18604305.bam
Perintah-perintah di atas merupakan perintah dasar, dan Anda dapat menggunakan teknik looping sehingga proses alignment dan konversi dapat berjalan secara simultan untuk semua sampel dengan cara sebagai berikut:
# bash
# buat direktori untuk hasil alignment dan file .bam
mkdir -p HISAT-StringTie/aligned_data
mkdir -p HISAT-StringTie/bam_data
# daftar nama sampel
samples=(
"SRR18604276" "SRR18604277" "SRR18604278" "SRR18604279"
"SRR18604280" "SRR18604281" "SRR18604282" "SRR18604283"
"SRR18604284" "SRR18604285" "SRR18604286" "SRR18604287"
"SRR18604288" "SRR18604289" "SRR18604290" "SRR18604291"
"SRR18604292" "SRR18604293" "SRR18604294" "SRR18604295"
"SRR18604296" "SRR18604297" "SRR18604298" "SRR18604299"
"SRR18604300" "SRR18604301" "SRR18604302" "SRR18604303"
"SRR18604304" "SRR18604305"
)
# loop untuk proses alignment dan konversi untuk setiap sampel
for sample in "${samples[@]}"; do
echo "Memproses sampel: $sample"
# Langkah 1: Alignment dengan HISAT2
hisat2 -p 4 --dta \
-x HISAT-StringTie/index/ASM2237912v1_hisat2_index \
--rna-strandness RF \
-1 raw_data/${sample}_1.fastq.gz \
-2 raw_data/${sample}_2.fastq.gz \
-S HISAT-StringTie/aligned_data/${sample}.sam
# Langkah 2: Konversi SAM ke BAM
samtools view -@ 6 -bS HISAT-StringTie/aligned_data/${sample}.sam > HISAT-StringTie/bam_data/${sample}.bam
# Langkah 3: Sorting BAM file
samtools sort -@ 6 HISAT-StringTie/bam_data/${sample}.bam -o HISAT-StringTie/bam_data/${sample}.sorted.bam
# Langkah 4: Indexing BAM file
samtools index HISAT-StringTie/bam_data/${sample}.sorted.bam
# Langkah 5: Hapus file SAM dan BAM mentah untuk menghemat ruang
rm HISAT-StringTie/aligned_data/${sample}.sam
rm HISAT-StringTie/bam_data/${sample}.bam
echo "Selesai memproses $sample"
echo "------------------------------------------"
done
Merakit transkrip dan menghitung level ekspresi gen menggunakan StringTie
Untuk merakit (assembling) transkrip RNA dan menghitung abundansi/ekspresi gen dari data RNA sequencing yang sudah disejajarkan (alignment), kita akan menggunakan StringTie dan menggunakan input data untuk semua sampel dengan format .sorted.bam dan file anotasi references/GCF_042767895.1_ASM4276789v1_genomic.gff.
Perintah dasar StringTie untuk assemby dan kuantifikasi adalah sebagai berikut:
# bash
# membuat direktori output
mkdir -p HISAT-StringTie/stringtie_output/
# menjalankan stringtie
stringtie HISAT-StringTie/bam_data/nama_sampel.sorted.bam \
-p 4 \
-G references/GCF_042767895.1_ASM4276789v1_genomic.gff \
--rf \
-A HISAT-StringTie/stringtie_output/nama_sampel_abundance.tab \
-o HISAT-StringTie/stringtie_output/nama_sampel.gtf \
-l nama_sampel
Dalam perintah di atas, ada beberapa parameter penting diantaranya:
-p: berapa banyak thread/core CPU yang ingin digunakan, misalnya 4, 6, 8, dst--rf: parameter untuk menentukan orientasi strand data sequencing (--rfuntuk stranded atau--fruntuk unstranded)-A: menghasilkan file abundansi/ekspresi dalam format tabular yang berisi informasi kuantitatif gen dan transkrip-o: menentukan file output utama berupa hasil assembly transkrip dalam format.gtf-l: memberikan label/prefix untuk penamaan transkrip yang dirakit oleh StringTie
Karena kita akan melakukan assembly dan kuantifikasi untuk semua sampel, maka teknik looping sangat diperlukan.
# bash
# membuat direktori output stringtie
mkdir -p HISAT-StringTie/stringtie_output/
# daftar nama sampel
samples=(
"SRR18604276" "SRR18604277" "SRR18604278" "SRR18604279"
"SRR18604280" "SRR18604281" "SRR18604282" "SRR18604283"
"SRR18604284" "SRR18604285" "SRR18604286" "SRR18604287"
"SRR18604288" "SRR18604289" "SRR18604290" "SRR18604291"
"SRR18604292" "SRR18604293" "SRR18604294" "SRR18604295"
"SRR18604296" "SRR18604297" "SRR18604298" "SRR18604299"
"SRR18604300" "SRR18604301" "SRR18604302" "SRR18604303"
"SRR18604304" "SRR18604305"
)
# menghitung total sampel dan inisialisasi counter
total=${#samples[@]}
counter=1
for sample in "${samples[@]}"; do
echo ""
echo "Running StringTie for sample: $sample ($counter/$total)"
echo "Started at: $(date)"
stringtie HISAT-StringTie/bam_data/${sample}.sorted.bam \
-p 4 \
-G references/GCF_042767895.1_ASM4276789v1_genomic.gff \
--rf \
-A HISAT-StringTie/stringtie_output/${sample}_abundance.tab \
-o HISAT-StringTie/stringtie_output/${sample}.gtf \
-l ${sample}
echo "Sample $sample StringTie completed at: $(date)"
echo "Progress: $counter/$total samples completed"
echo "--------------------------------------------------"
((counter++))
done
Menggabungkan transkrip dengan StringTie Merge
Setelah menjalankan StringTie untuk setiap sampel, langkah selanjutnya adalah menggabungkan semua file GTF yang dihasilkan menjadi satu set transkrip yang konsisten menggunakan perintah stringtie --merge. Ini penting untuk memastikan bahwa semua sampel menggunakan set transkrip yang sama untuk analisis kuantitatif yang akurat.
# bash
# membuat file daftar GTF untuk merge
echo "Membuat list file GTF untuk digabungkan..."
ls HISAT-StringTie/stringtie_output/*.gtf > HISAT-StringTie/stringtie_output/gtf_list.txt
# menjalankan stringtie merge
echo "Menggabungkan file gtf dengan stringtie..."
stringtie --merge \
-p 4 \
-G references/GCF_042767895.1_ASM4276789v1_genomic.gff \
-o HISAT-StringTie/stringtie_output/merged.gtf \
HISAT-StringTie/stringtie_output/gtf_list.txt
echo "Penggabungan selesai pada: $(date)"
echo "File GTF yang digabungkan disimpan di: HISAT-StringTie/stringtie_output/gtf_list.gtf"
Parameter yang digunakan dalam stringtie --merge:
--merge: mode untuk menggabungkan multiple GTF files-p: jumlah thread yang digunakan-G: file anotasi referensi-o: file output merged GTF- File terakhir adalah daftar file GTF yang akan digabungkan
Re-kuantifikasi dengan merged GTF
Setelah mendapatkan set transkrip yang sudah digabungkan, kita perlu menjalankan StringTie sekali lagi untuk setiap sampel menggunakan merged GTF sebagai referensi. Tahap ini akan menghasilkan data kuantitatif yang konsisten untuk semua sampel. Kita akan menggunakan teknik looping untuk semua sampel
# bash
# membuat direktori ballgown untuk hasil re-quantification
mkdir -p HISAT-StringTie/stringtie_output/ballgown
# daftar nama sampel
samples=(
"SRR18604276" "SRR18604277" "SRR18604278" "SRR18604279"
"SRR18604280" "SRR18604281" "SRR18604282" "SRR18604283"
"SRR18604284" "SRR18604285" "SRR18604286" "SRR18604287"
"SRR18604288" "SRR18604289" "SRR18604290" "SRR18604291"
"SRR18604292" "SRR18604293" "SRR18604294" "SRR18604295"
"SRR18604296" "SRR18604297" "SRR18604298" "SRR18604299"
"SRR18604300" "SRR18604301" "SRR18604302" "SRR18604303"
"SRR18604304" "SRR18604305"
)
# re-quantification untuk setiap sampel
counter=1
for sample in "${samples[@]}"; do
echo "Re-estimating for sample: $sample ($counter/$total) at: $(date)"
mkdir -p HISAT-StringTie/stringtie_output/ballgown/${sample}
stringtie HISAT-StringTie/bam_data/${sample}.sorted.bam \
-e -B \
-p 4 \
--rf \
-G HISAT-StringTie/stringtie_output/merged.gtf \
-o HISAT-StringTie/stringtie_output/ballgown/${sample}/${sample}.gtf
if [ $? -eq 0 ]; then
echo "Sample $sample re-estimation completed successfully"
else
echo "ERROR: Re-estimation failed for sample $sample"
fi
((counter++))
done
Parameter yang digunakan dalam re-estimasi:
-e: hanya mengestimasi abundansi transkrip yang sudah ada di merged GTF-B: menghasilkan file Ballgown untuk analisis downstream-G: menggunakan merged GTF sebagai referensi
Preparasi data untuk analisis downstream dengan prepDE.py
Tahap terakhir adalah menyiapkan data dalam format count matrix untuk analisis ekspresi diferensial menggunakan script prepDE.py dari StringTie.
# bash
# download prepDE.py jika belum ada
if [ ! -f "prepDE.py" ]; then
echo "Menunduh prepDE.py..."
wget https://ccb.jhu.edu/software/stringtie/dl/prepDE.py
chmod +x prepDE.py
fi
# membuat file sample list untuk prepDE.py
echo "Membuat list sampel untuk prepDE.py..."
echo -e "sample_id\tpath" > HISAT-StringTie/sample_list.txt
for sample in "${samples[@]}"; do
echo -e "${sample}\tHISAT-StringTie/stringtie_output/ballgown/${sample}/${sample}.gtf" >> HISAT-StringTie/sample_list.txt
done
# menjalankan prepDE.py
echo "Menjalankan prepDE.py untuk mengekstrak count matrix..."
python prepDE.py -i HISAT-StringTie/sample_list.txt \
-g HISAT-StringTie/gene_count_matrix.csv \
-t HISAT-StringTie/transcript_count_matrix.csv
echo "Hasil:"
echo "- Gene count matrix: HISAT-StringTie/gene_count_matrix.csv"
echo "- Transcript count matrix: HISAT-StringTie/transcript_count_matrix.csv"
echo "Selesai pada: $(date)"
Kode di atas adalah kode dasar untuk memperoleh data siap analisis differential expression. Tapi sering kali kita akan menjumpai ketidaksesuaian antara file GTF hasil StringTie dan file count data (t_data.ctab) yang dihasilkan oleh parameter -B (Ballgown). Masalah ini dapat menyebabkan hilangnya beberapa transkrip dalam analisis downstream.
Secara umum, untuk mengatasi ini kita perlu:
- Memvalidasi konsistensi data antara file GTF dan file Ballgown: Gunakan
grepdancommuntuk membandingkan transcript IDs antara file GTF dant_data.ctab - Jika ditemukan ketidaksesuaian, kita dapat memperbaiki file GTF dengan menambahkan transkrip yang hilang: Gunakan
cut,echo -e, dan redirection (>>) untuk mengekstrak koordinat darit_data.ctabdan menambahkan entry baru ke file GTF. - Regenerasi count matrix dengan data yang sudah diperbaiki: Ulangi
prepDE.pyuntuk menghasilkan count matrix yang lengkap dan konsisten.
Mengingat mulai dari proses aligment hingga kuantifikasi cukup kompleks, saya telah menyiapkan bash script untuk otomatisasi semua jenis data sequencing, selama data itu adalah paired-end reads dan stranded. Download kedua file config.yaml dan All-hisat-stringtie.sh.
Anda hanya perlu mengatur file config.yaml, simpan, dan jalankan script All-hisat-stringtie.sh dengan perintah bash All-hisat-stringtie.sh.